Software
Computational chemists
Meaningful computational results that inform molecule design
Enhance your designs using advanced approaches for protein-ligand analysis
Flare™ is an agile ligand- and structure-based drug design solution that enables you to discover novel small molecules more efficiently and effectively in a single platform.
- Calculate relative binding affinity within a congeneric ligand series using Flare FEP for robust and efficient calculations and accurate ligand-binding affinities
- Inform new molecule design with electrostatics and get visual feedback on new molecule designs to understand ligand binding, structure-activity relationships and rank new molecule designs
- Get detailed feedback on your molecule designs with excellent pose predictions, rapidly and easily dock to multiple protein conformations in a single experiment, and easily predict the binding pose and interactions of covalent inhibitors
- Study the conformational changes of proteins and assess the stability of protein-ligand complexes with Molecular Dynamics using OpenMM
- Understand which water molecules are tightly bound to the active site of the protein and which are energetically unfavorable using 3D-RISM and GIST analysis
- Customize and automate your tasks using the Python API which enables you to create your own workflows, automate common tasks, expand Flare with Python modules and add custom controls, for more efficient working
Increase your wet screening hit rate at a fraction of the cost
Blaze™ enables you to make efficient use of your time by routinely running a virtual screen in parallel to wet screening. A virtual screen of 10 million structures just takes a few hours.
Customers have been using Blaze to generate lead-like hits for over a decade, achieving hit rates as high as 30%, but what they really value is the diversity of hits returned from every Blaze screen.
Facilitate communication and exchange of ideas
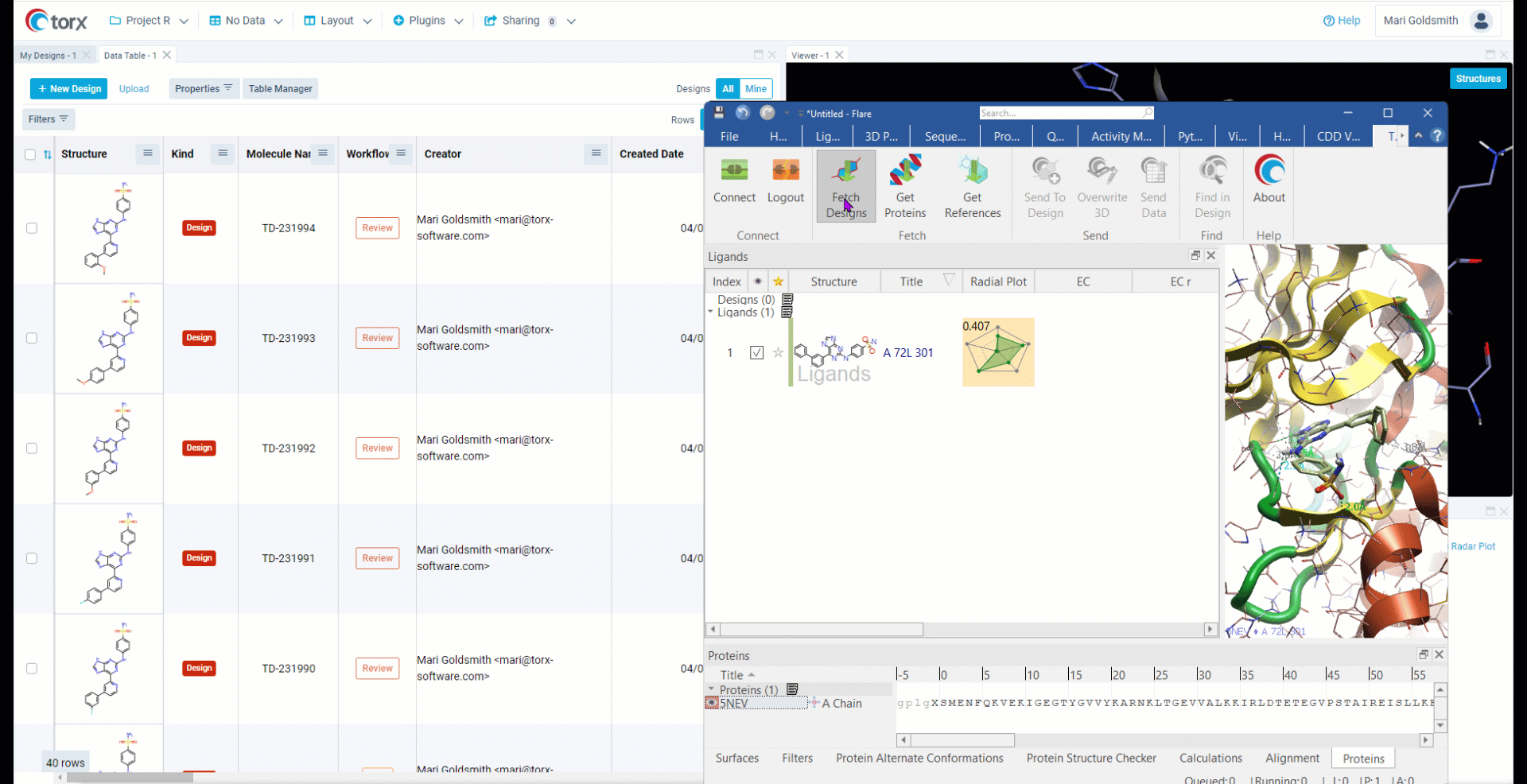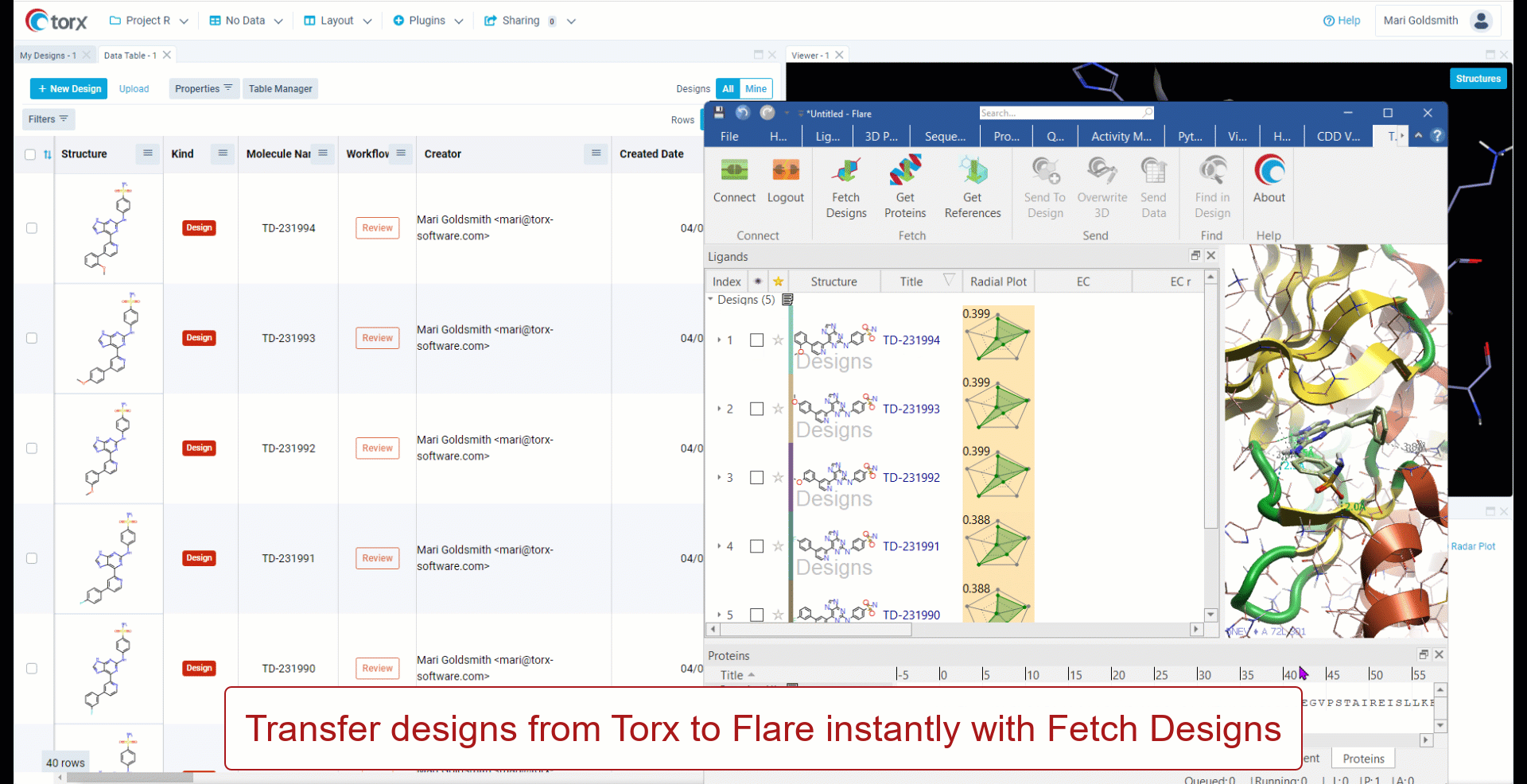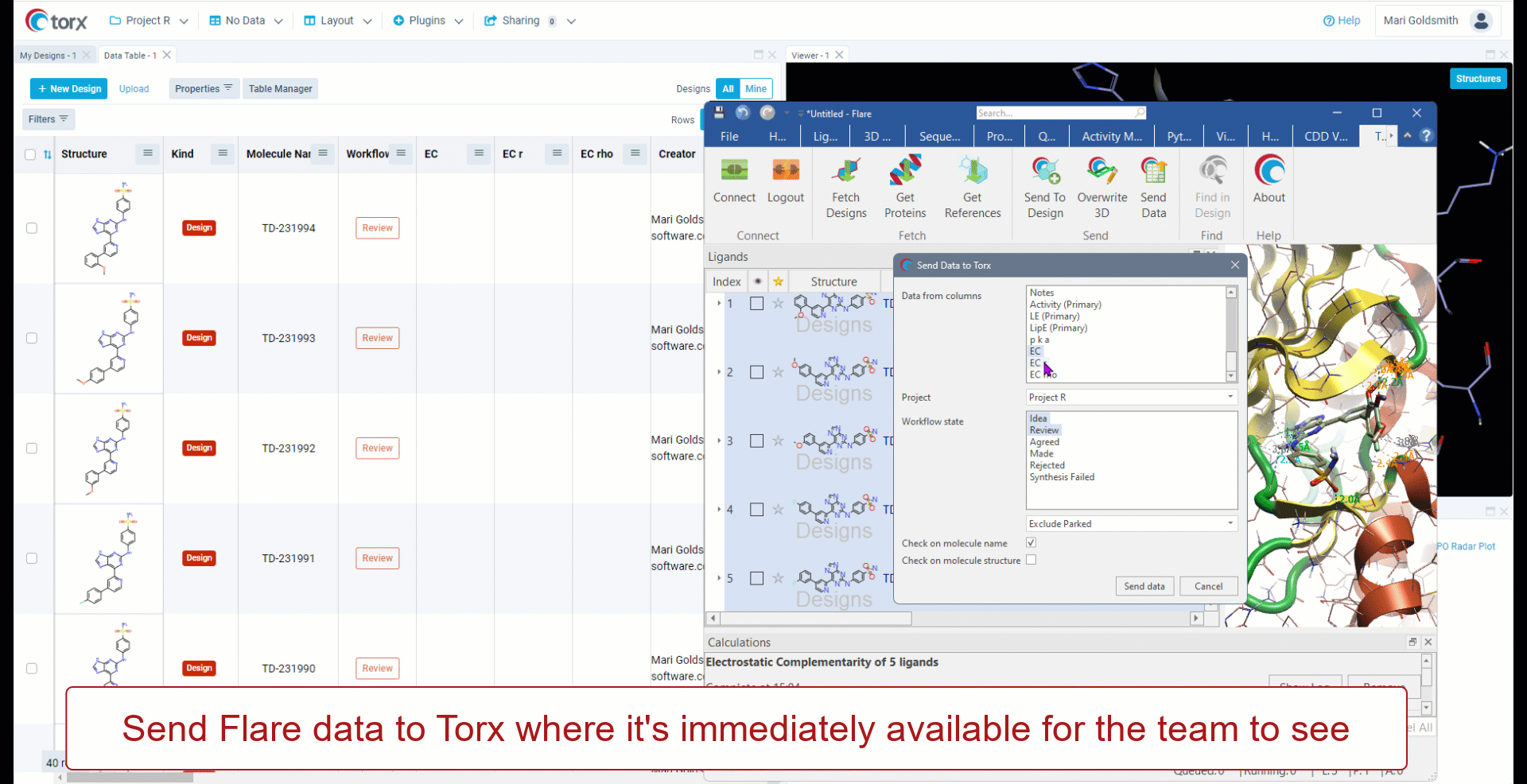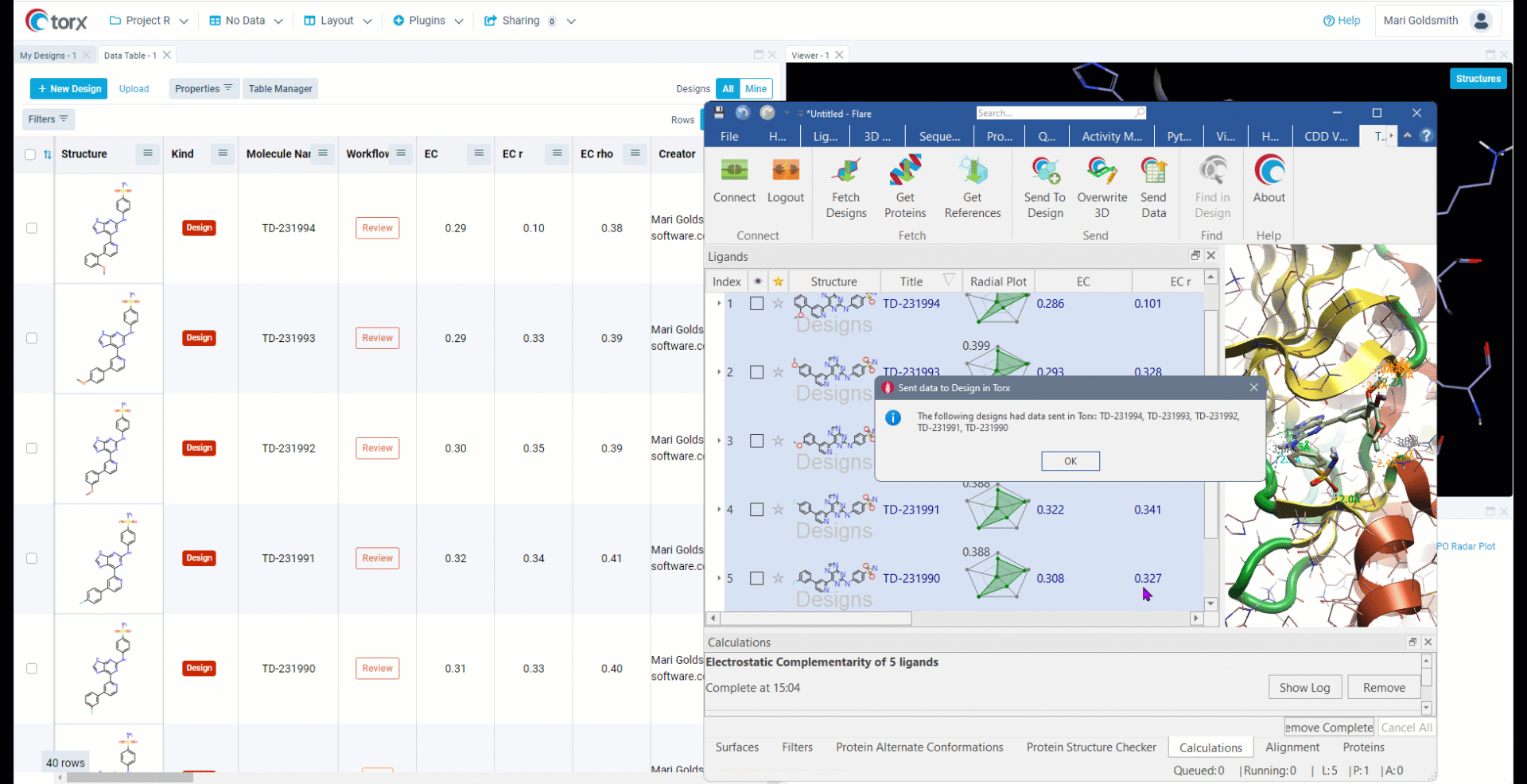
Cresset has partnered with Elixir Software to deliver Torx® - a visual, chemistry aware, cloud-based DMTA (design-make-test-analyze) platform. Torx also offers an out-of-the-box integration to Flare, allowing users to fetch compounds, proteins and reference ligands to perform experiments such as docking and FEP (free energy perturbation).
- Simple integration with Torx Design
- Connect to and transfer information from Flare using simple API extension
- Facilitate communication with medicinal chemists
- Real-time exchange of ideas and hypotheses