flare™
Docking many ways
Rapidly and easily dock your ligands from a choice of different experiments
Flare Docking uses Lead Finder™ to give you detailed feedback on your new molecule designs, high enrichments in virtual screening and excellent pose prediction.
Use Flare Docking to tackle the flexibility of the protein active site with ensemble docking run on multiple protein conformations, and easily predict the binding pose and interactions of covalent inhibitors choosing your preferred covalent warhead.
Request recording of webinar 'Streamline the design of covalent inhibitors'
- Predict the 3D structure of non-covalently bound protein-ligand complexes by docking a flexible ligand to a static protein structure
- Dock covalent ligands known to bind to a particular residue in the protein, using a variety of reactive warheads
- Dock covalent and non-covalent ligands to multiple protein structures in one experiment
- Use a ligand template to seed a docking of multiple ligands that share a common substructure, ultimately leading to better docking results
- Set docking constraints to bias results to include specific protein-ligand interactions, such as H-bonds, protein metals, pi-stacking, pi-cation, or salt-bridges
- Dock hundreds of compounds in parallel using Cresset Engine Broker™
- Rapidly assess new molecule designs for their fit to the protein active site
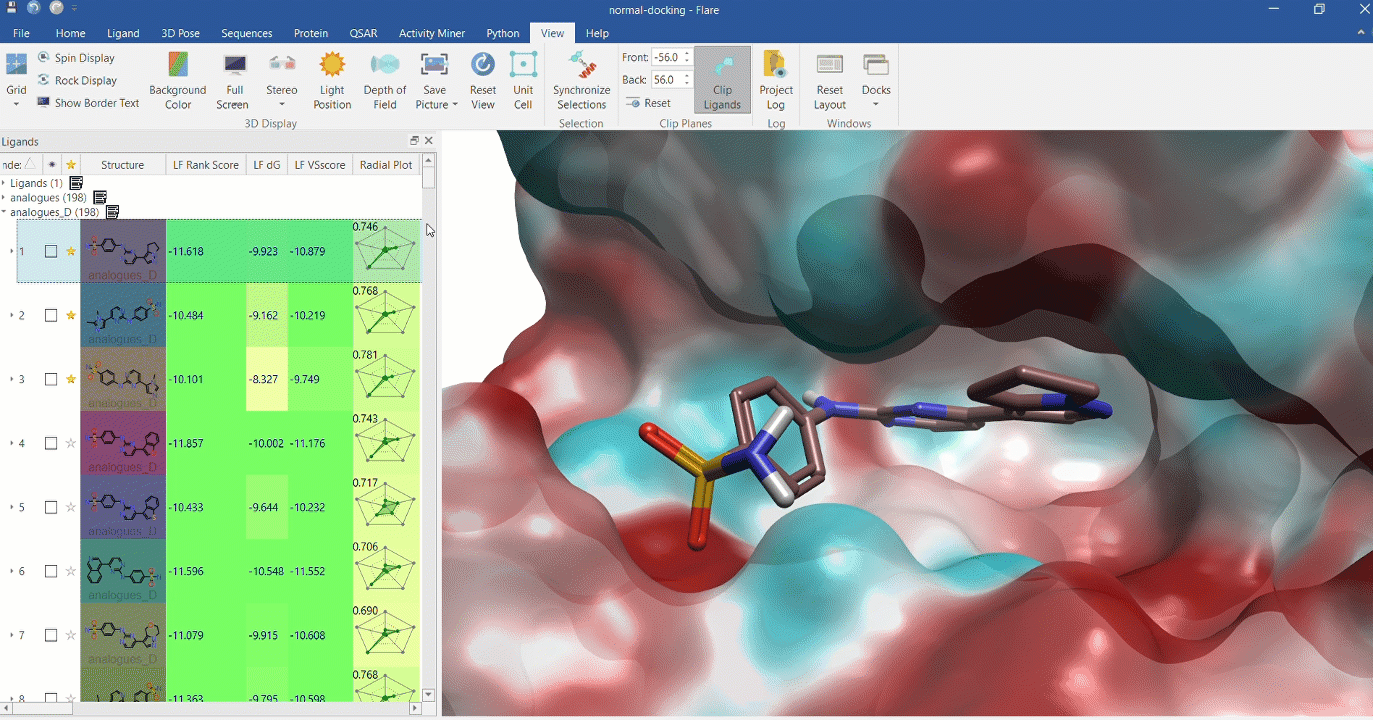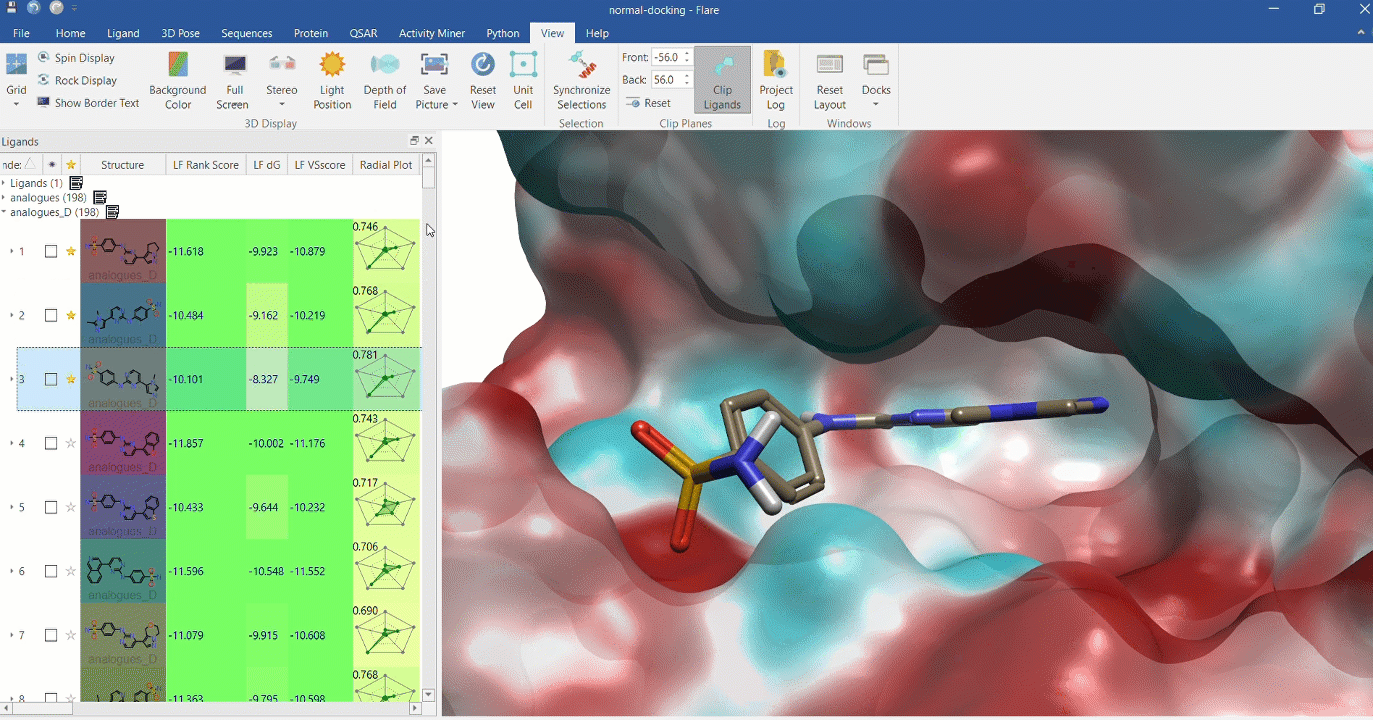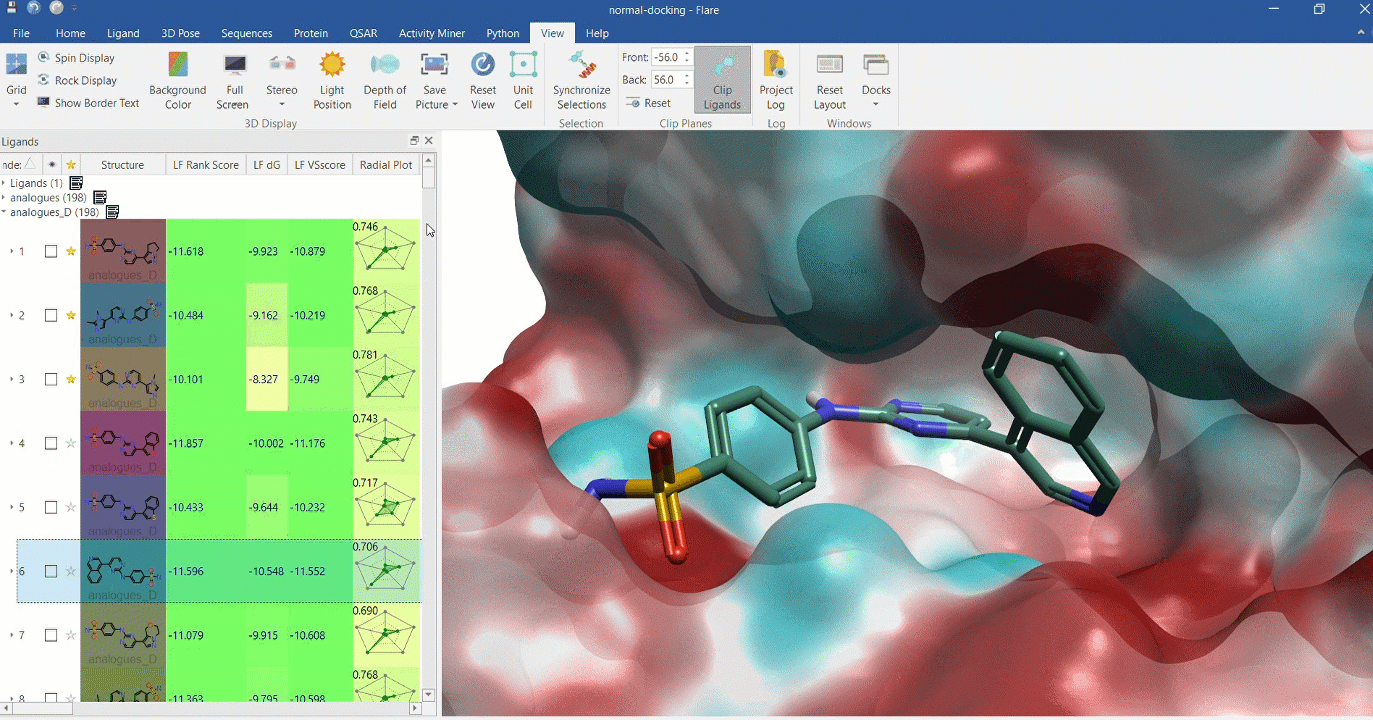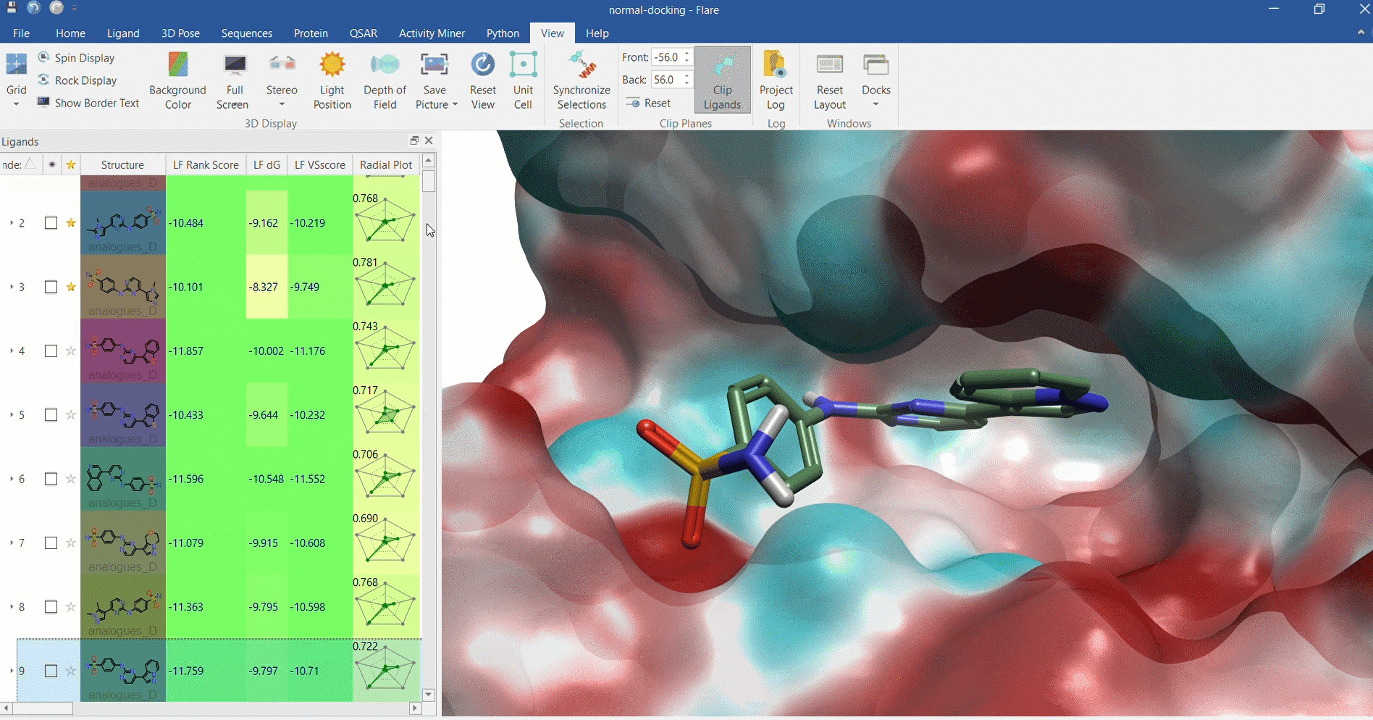
Docking multiple ligands to a single protein
Multiple analogues of the crystallographic ligand in PDB:1OIT docked into the protein's active site. The surface of the 1OIT active site is colored by electrostatic potential (red = positive, blue = negative).
Docking covalent inhibitors
Docking of a covalent inhibitor targeting Tyrosine, displayed with Electrostatic Complementary™ surface of PDB 4QDE
Docking to multiple proteins
Docking to multiple protein structures in a single experiment with concatenated results, displayed with the electrostatic surface of PDB 5HLW. Available for non-covalent and covalent ligands.