flare™
Molecular Dynamics
Study the conformational changes of proteins and assess the stability of protein-ligand complexes
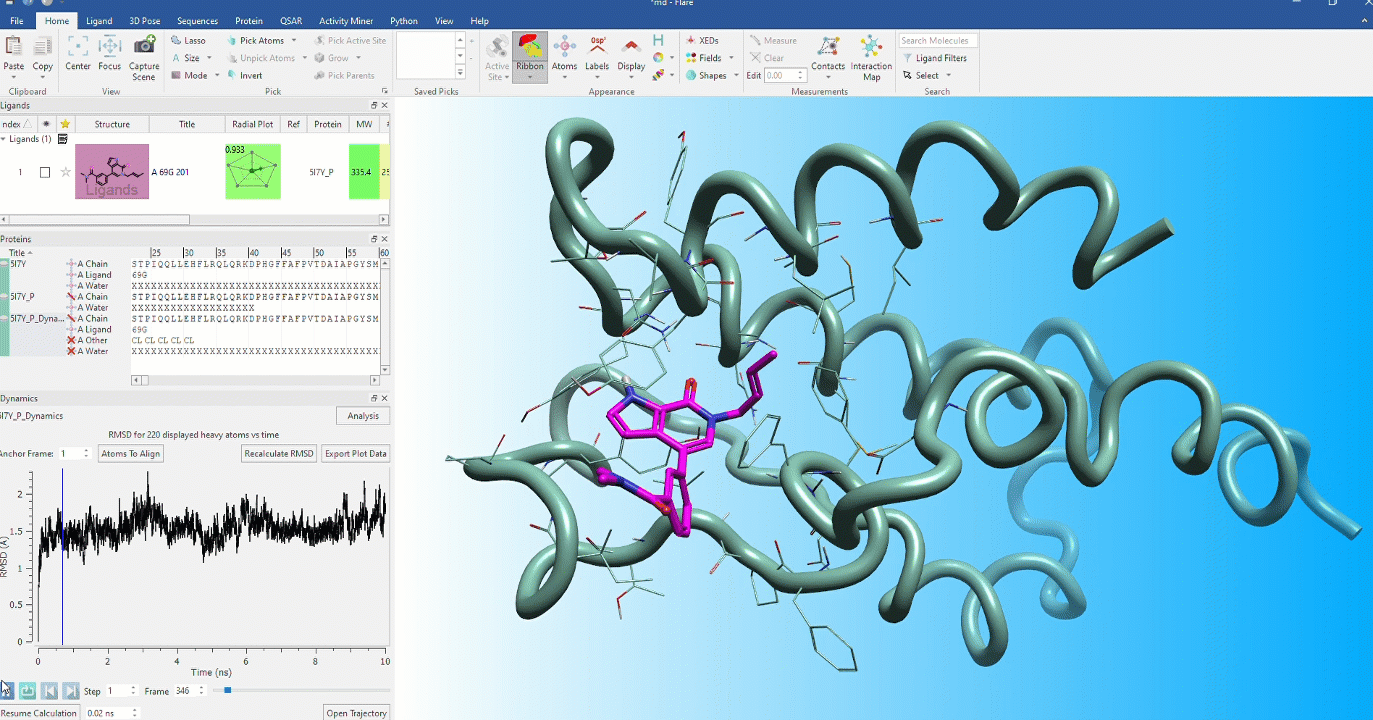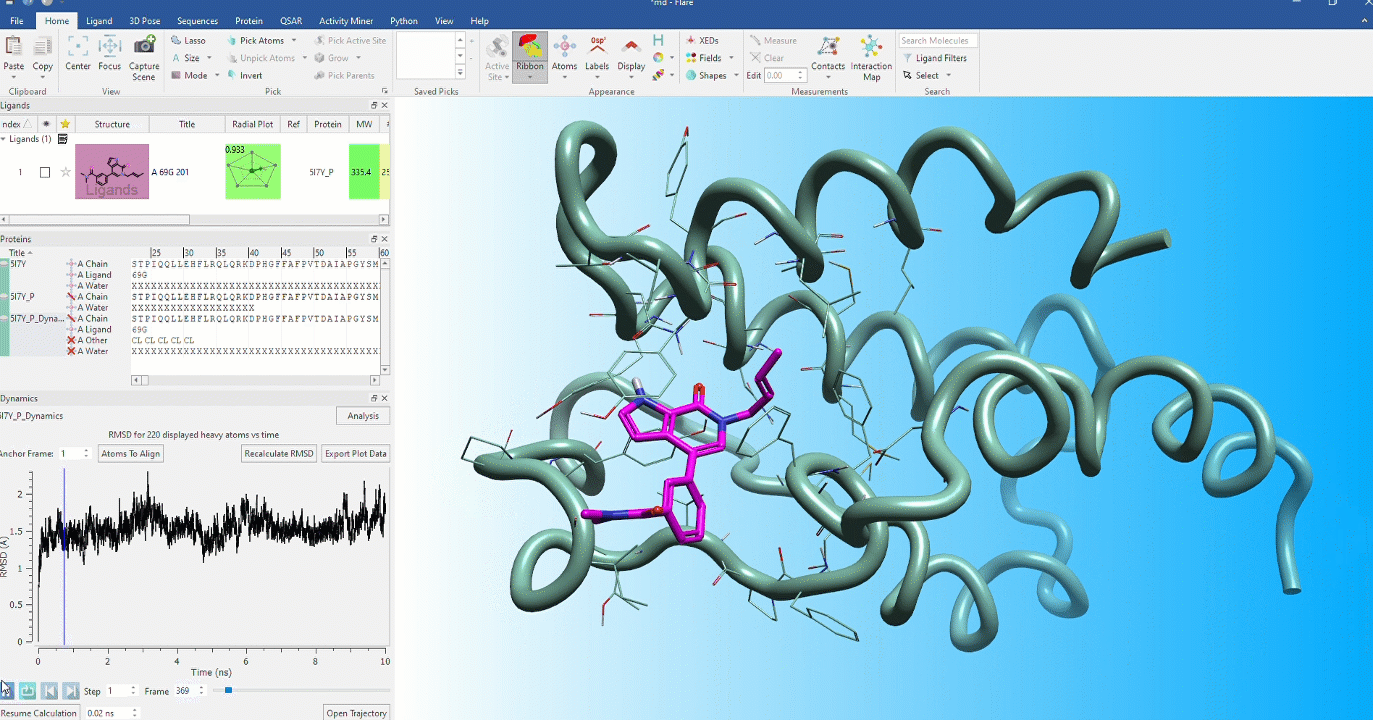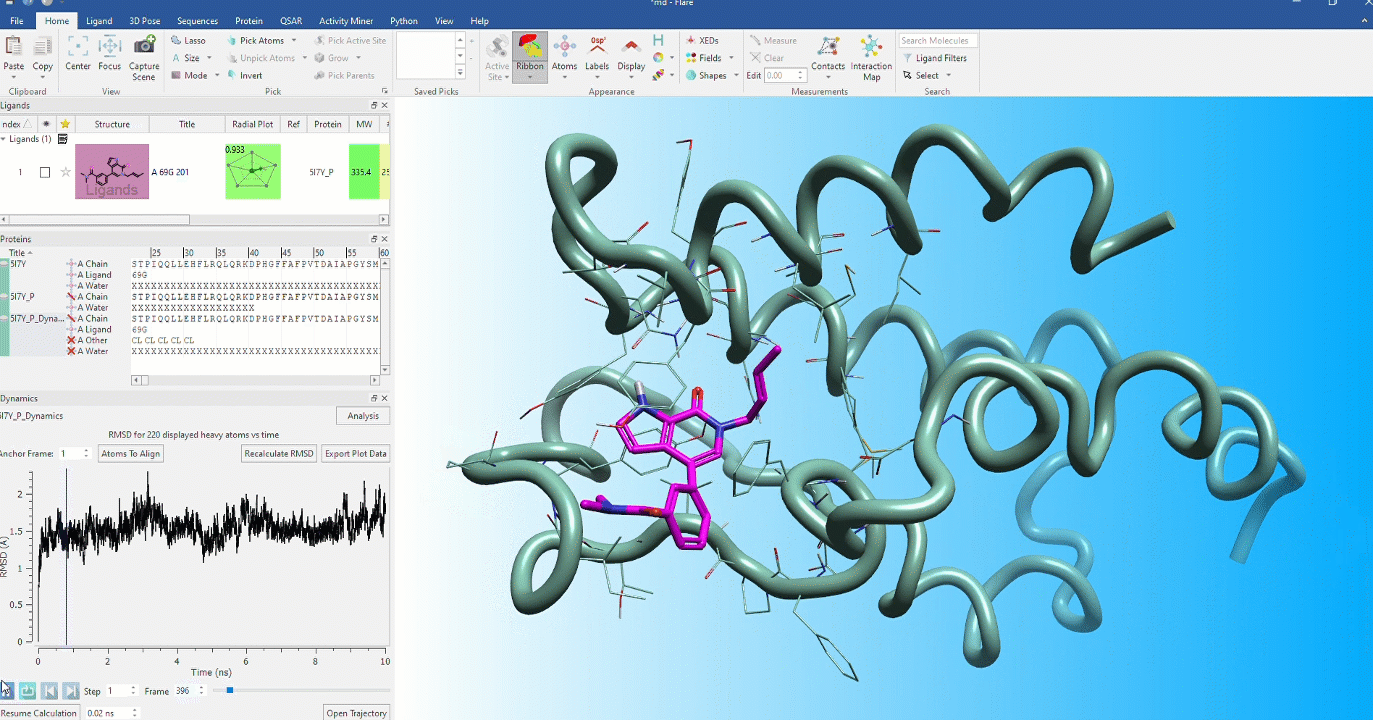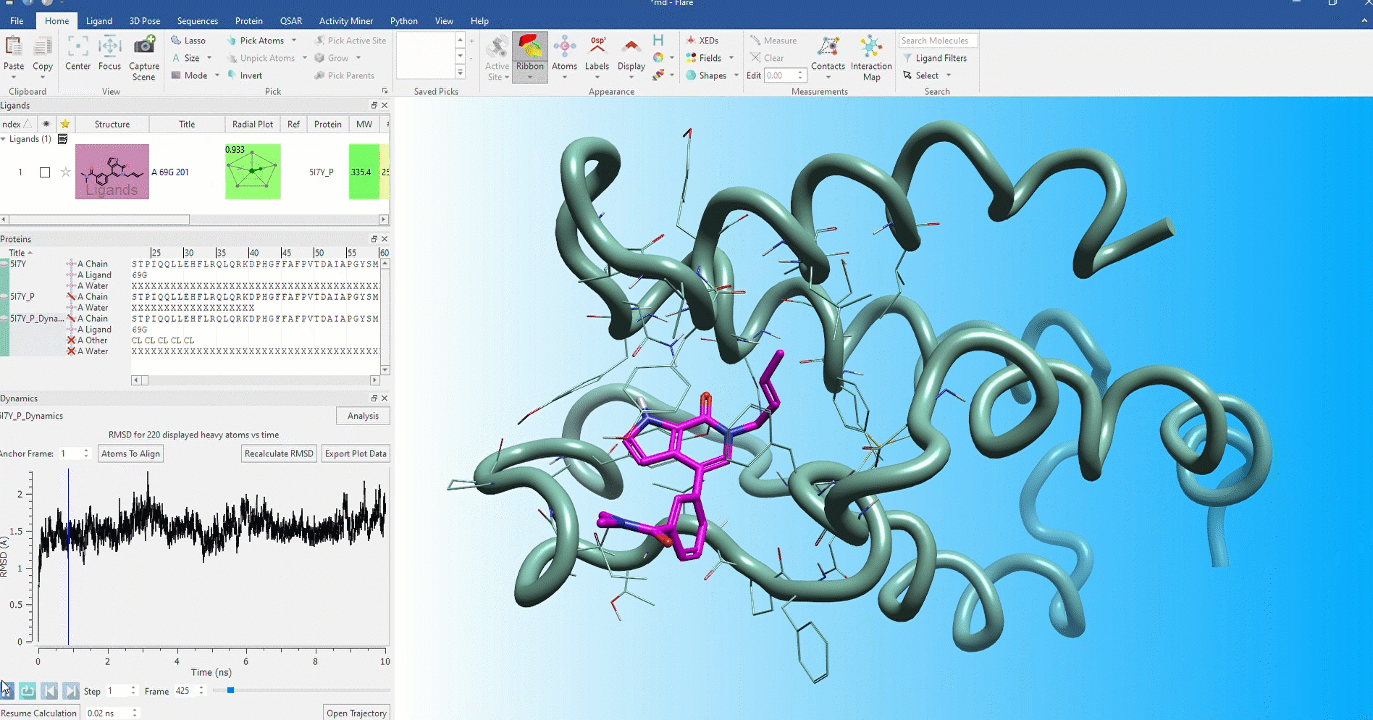
Dynamic stability of protein conformations
The dynamic stability of protein conformations has become a key focus in the study of proteins and protein-ligand complexes. Flare provides a dedicated interface to create, playback and analyze Molecular Dynamics trajectories, based on OpenMM.
- Study docking results to determine pose stability through contact frequency analysis
- View initial ligand pose during the trajectory playback to visually assess ligand movements and pose changes
- Easily take snapshots as starting points for further experiments, such as ensemble docking or FEP calculations
Bespoke and rapid setup of experiments
Flare enables highly customized or standard configurations for calculations enabling both bespoke and rapid setup of experiments. Key features in dynamics calculations are:
- Multiple force field support – choose AMBER (GAFF, GAFF2) or the Open Force Field
- Enhance the accuracy of your results by automatically creating custom torsion parameters for small molecules using GFN2-xTB extended semiempirical tight-binding, ANI-2x deep learning QM approximation or or hybrid DFT//GFN2-xTB calculations
- Choose explicit water models with three of four point models for the greatest accuracy
- Run simulations of membrane proteins choosing between two types of lipids (POPC,POPE) to create the bilayer
- Enhance water sampling with Grand Canonical Nonequilibrium Candidate Monte Carlo (GCNCMC)
- Run longer simulations in a shorter time using Hydrogen Mass Repartitioning and longer time steps
- Use either local GPUs/CPUs to get the most out of your hardware, or rely upon the seamless connection to remote calculation resources offered by the Cresset Engine Broker™ to speed up the calculation
Analysis of dynamics trajectories in Flare
The analysis of dynamics trajectories in Flare provides the feedback you need to make decisions.
- Dedicated dynamics dock enables visualization of trajectories in the context of your complete Flare project enabling comparison with starting protein and ligand conformations
- Analyze dynamics trajectories with informative, interactive visual tools such as RMSD, 2D RMSD and RMSF plots and secondary structure analysis tables
- Analyze protein-ligand interactions and custom measurements across all or part of the trajectory
- Cluster the dynamics trajectory and select representative centroid conformations to use as starting points for further experiments
- Gain confidence in your results by analyzing system physical properties like potential energy, temperature, box volume and density
- Flare uses the powerful OpenMM dynamics engine enabling saving and reading of trajectories in standard DCD format wherever they are calculated
References and acknowledgements
P. Eastman, J. Swails, J. D. Chodera, R. T. McGibbon, Y. Zhao, K. A. Beauchamp, L.-P. Wang, A. C. Simmonett, M. P. Harrigan, C. D. Stern, R. P. Wiewiora, B. R. Brooks, and V. S. Pande, OpenMM 7: Rapid development of high performance algorithms for molecular dynamics, PLOS Comp. Biol. 2017, 3(7): e1005659
M. L. Samways, H. E. Bruce Macdonald, J. W. Essex, grand: A Python Module for Grand Canonical Water Sampling in OpenMM, J. Chem. Inf. Model. 2020, 60, 10, 4436-4441
O. J. Melling, M. L. Samways, Y. Ge, D. L. Mobley, J. W. Essex, Enhanced Grand Canonical Sampling of Occluded Water Sites Using Nonequilibrium Candidate Monte Carlo, J. Chem. Theory Comput. 2023, 19, 1050-1062
C. Bannwarth, S. Ehlert, S. Grimme, GFN2-xTB—An accurate and broadly parametrized self-consistent tight-binding quantum chemical method with multipole electrostatics and density-dependent dispersion contributions, J. Chem. Theory Comp. 2019, 15, 3, 1652-1671
C. Devereux, J. S. Smith, K. K. Huddleston, K. Barros, R. Zubatyuk, O. Isayev, and A. E. Roitberg, Extending the applicability of the ANI deep learning molecular potential to sulfur and halogens, J. Chem. Theory Comput. 2020, 16,7, 4192–4202
P. K. Behara, H. Jang, J. Horton, D. Dotson, S. Boothroyd, C. Cavender, V. Gapsys, T. Gokey, D. Hahn, J. Maat, O. Madin, I. Pulido, M. Thompson, J. Wagner, L. Wang, J. Chodera, D. Cole, M. Gilson, M. Shirts, C. Bayly, L.-P. Wang, D. Mobley, Benchmarking QM theory for drug-like molecules to train force fields, Poster presented at CUP XXI, March 08, 2022 - March 10, 2022, Santa Fe, New Mexico, USA